

الضُّمور -الحَثل- العَضَلي
الضُّمور -الحثل- العضليّ (The muscular dystrophies MD) هو حالة وراثيَّة متقدمة تُحدِث ضعفًا في العضلات، مما ينتُج عنه إعاقة تتمثَّل في فُقدان تدريجيّ للقُدرة على المشي والكَلام والعناية بالنَّفس ومن ثم المَوت.
يُعَدُّ ضمُور العضلات (MD) مرضًا نادر الحُدوث، ويختلف من مريض لآخر ممن يحدث لهم الخلل الجيني نفسُه، كما يختلف وقت ظهور الأعراض وشدتها من شخصٍ لآخر أيضًا، وبالتالي قد تختلف الخُطة العلاجية، لذلك من الصَّعب التنبؤ بمسيرته. [1،2]
انتشار مرض الضمُّور العضلي
تُقدَّر نسبة انتشار المرض في مصر طبقًا لما ذكره الباحث الطلاوي من جامعة أسيوط عام 2005 بـ 26.8 بين 100 ألف شخص، وهي نسبة تتوافَق مع مُعظم الدراسات التي أُجريت في البلدان الأخرى حول العالم. [1]
أسباب مرض الضمُّور العضلي
مرض الضمور العضلي (MD) مرضٌ وراثيّ، نتيجة توريث طفرات (Mutations) للجين المسؤول عن تكوين البروتين الخاص ببناء العضلات، ومن النادر جدًا حدوثه بخلاف ذلك. ينتج عن هذه الطفرة بروتينات بكميات قليلة أو بأنواع غير قادرة على أداء الوظيفة المُسنَدة إليها، والذي بدوره يؤثر على تركيب ووظيفة الألياف العضليّة. [1]
أنواع الضمُّور العضليّ
يتمثل الضمور العضلي (MD) في مجموعة من الأمراض التي تظهر على هيئة ضعف في العضلات، وتغيُّر في تركيب الخلايا، الذي ينتج عنه تغير في البنيان العضليّ وفقدان النسيج السليم.
في بعض الحالات لا يقتصر المرض على العضلات فقط، وإنما يمتد الضرر ليشمل أعضاءً أخرى مثل القلب. [3]
قُسِّم الضمور العضلي -حتى وقت قريب- علميًا إلى ستة أصناف تتمثل في:
- ضمور دوشين وبيكر العضلي (Duchenne and Becker MD)
- الحثل العضلي في الحزام الطرفي (Limb-girdle MD)
- اعتلال عضلي قاص (Distal Myopathies)
- ضمور عضلي وراثي (Congenital MD)
- الحثل العضلي الوجهي الكتفي العضدي (Facio-Scapulo-humeral MD)
- الحثل التأتُّري العضلي (Myotonic MD)
أعراض مرض الضمُّور العضلي
يتنوع وقت بدء الأعراض وظهورها في أمراض الضمور العضلي من الولادة حتى سن البلوغ، فالنوع الورَاثي (The congenital muscular dystrophies) يظهر منذ الولادة أو خلال الشهور الأولى من الحياة، أما دوشين (Duchenne MD) وحثل الحزام الطرفي (Limb-girdle MD) يظهران في سن الطفولة المبكرة أو خلال فترة المراهقة.
ضعف العضلات الهيكليّة (Skeletal muscle) يميِّز كل أنواع الضمور العضلي، ومع ذلك تختلف شدة وطبيعة المرض والأجزاء التي يشملها هذا الضعف بين الأنواع المختلفة، مع الحاجة لكرسي متحرك (wheelchair) في نهاية المطاف.
يظهر الضعف العضليّ لدى الأطفال على هيئة:
- سقوط متكرر.
- عدم القدرة على النهوض، صعود الدرج، الجري أو القفز.
- المشي على أطراف الأصابع مشية مترنحة.
- انحناء العمود الفقري.
- تدلي الجفون.
- مشاكل في التنفس أو البلع.
- مشاكل في الرؤية.
توجد علامات أخرى تظهر لدى هذا المريض إضافة لضمور العضلات، فقد يحدث تضخُّم (Compensatory hypertrophy) لها في محاولة لتعويض الضعف الحاصل، تقلص المفاصل (Joint contractures)، توتر عضلي (Myotonia) ثم إصابة الجهاز التنفسي والقلب ببعض التغيرات التي تؤثر على وظيفتهم.
خلل الجهاز التنفسي (Respiratory impairment) لهو أمر شائع لدى مرضى الضمور العضلي باختلاف وقت الحدوث، ودرجة الخلل من مريض لآخر، بشكل عام يظهر تأثر الجهاز التنفسي بعد فقدان المريض القدرة على المشي، ويعزى ذلك إلى ضعف في عملية الشهيق وما يصاحبه من ضعف في عضلة الحجاب الحاجز، إضافة إلى ضعف عملية الزفير، والعضلات المتحكمة بالسعال والبلع التي من واجبها طرد الإفرازات من مجرى النفس مما يؤدي لتراكمها، الأمر الذي يحتاج لتدخلٍ خارجيّ.
غالبًا ما يبدأ خلل الجهاز التنفسي في الليل، ثم يتطور ليشمل الليل والنهار على حد سواء على مدار شهور أو سنوات نظرًا لطبيعة المرض بطيئة التقدم. [1،2]
العمر المُتوقَّع للمريض وسبب الوفاة
معظم المصابين بالضمور العضلي يموتون في سن يقارب العشرين، نتيجةً لفشل الجهاز التنفسي أو نتيجة مضاعفات قلبية، وتتعدد أسباب الوفاة لدى المرضى وخاصة من لديهم تأثر في وظيفة الجهاز التنفسي وتتلخص في الآتي [2، 4]:
- الانخماص (Atelectasis)
- انسداد مجرى الهواء بالإفرازات المختلفة (Mucus plugging)
- التهاب رئوي (Pneumonia)
- ضيق وصعوبة في التنفس (Dyspnea)
- فشل الجهاز التنفسي (Respiratory failure)
تشخيص مرض الضمُّور العضلي
يتضمن التشخيص كل أو بعض المراحل الآتية:
- التاريخ المرضي، خاصة أين ومتى بدأ الضعف العضلي، وهل شمل أجزاء أخرى كالقلب والجهاز العصبي.
- التاريخ المرضي للعائلة.
- تحاليل مخبرية كقياس مستوى الكرياتين كاينيز (CK) في الدم.
- فحوصات كهربية للأعصاب والعضلات.
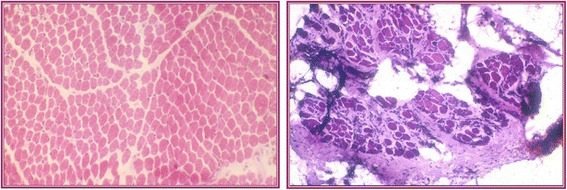
- عينة عضلية (Muscle biopsy) وفحص الأنسجة بالميكروسكوب (Histopathological analysis) لتحديد حجم وهيئة وكيفيّة تحلُّل وتجدد أو تليُّف النسيج العضلي.
- الاختبار الجيني (Genetic testing) ضروري لتحديد قابلية المريض للاستجابة للخطط العلاجية المختلفة. [5]
علاج مرض الضمُّور العضلي
حتى الآن لا يوجد علاج شافٍ لضمور العضلات، ولكن هناك العديد من الإجراءات المُتاحة التي يمكنها التحسين من الإعاقة الجسدية والمضاعفات المترتبة على هذا المرض.
تُركز الأبحاث في الوقت الحالي على اكتشاف كيفية تطور الجين المسبب للمرض، كما تُبذَل جهود عدة أيضًا لتطوير العلاج الخلوي (Cell-based therapeutic approaches) من أجل العثور على طريقة مُثلى لشفاء مرض ضمور العضلات.
يشمل العلاج الجيني (Gene therapy) توصيل الجين الناقص للعضلات المتأثرة بالمرض من أجل إنقاذ وظيفة البروتين، أما العلاج الخلوي (Cell therapy) فيركز على استبدال النسيج العضلي المُعتَّل بخلايا جذعية (stem/progenitor cells) قادرة على خلق أنسجة عضلية جديدة وسليمة، إضافة إلى دمج الأنسجة المتجددة بهدف استعادة وظيفة العضلات. كل من العلاج الجيني والخلوي يحمل تطلعات كبيرة في المستقبل.
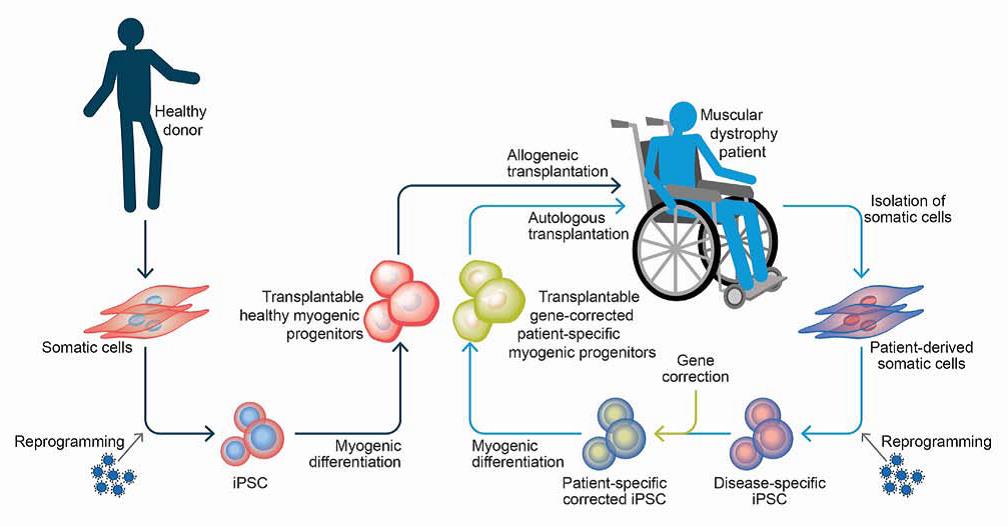
تمثل الخلايا الجذعية متعددة القدرات ((Pluripotent stem cells (PSCs) مصدرًا خصبًا للخلايا من أجل علاج الضمور العضلي، وذلك لقدرتها غير المحدودة على الانقسام والتميُّز لجميع أنواع الخلايا المكوُّنة للجسم، فهي تسمح بإنتاج أعداد ضخمة من الخلايا المولِّدة للخلايا العضلية بسهولة.
الخلايا الجذعية مُعادة البرمجة (Reprogramming technology PSCs) قد استُخدِمت في علاج العديد من أنواع الضمور العضلي، كما سمح التعديل الجيني (Genome editing) بتصحيح الطفرات الوراثيّة، مما فتح الباب أمام استخدامها في عمليات الزرع الذاتيّ. [1،6]
المصادر:
1. El-Tallawy HN, Khedr EM, Qayed MH, Helliwell TR, Kamel NF. Epidemiological study of muscular disorders in Assiut, Egypt. Neuroepidemiology. 2005;25(4):205–11.
2. Carter, J. C., Sheehan, D. W., Prochoroff, A., & Birnkrant, D. J. (2018). Muscular dystrophies. Clinics in chest medicine, 39(2), 377-389.
3. Falsaperla, R., Praticò, A. D., Ruggieri, M., Parano, E., Rizzo, R., Corsello, G., … & Pavone, P. (2016). Congenital muscular dystrophy: from muscle to brain. Italian journal of pediatrics, 42(1), 78.
4. Teymoori, A., Azimi-Nezhad, M., & Ebrahimzadeh-Vesal, R. (2018). A case report of Duchenne muscular dystrophy; identification of a novel mutation in dystrophin gene using next generation sequencing. Meta Gene, 18, 112-114.
5. Shieh, P. B. (2013). Muscular dystrophies and other genetic myopathies. Neurologic clinics, 31(4), 1009-1029.Chicago
6. Selvaraj, S., Kyba, M., & Perlingeiro, R. C. (2019). Pluripotent Stem Cell-Based Therapeutics for Muscular Dystrophies. Trends in molecular medicine.
إعداد: نهى محمود / أحمد منتصر
مراجعة علمية: أحمد منتصر
تَدقيقٌ لُغَوِيّ: محمود خليفة
تحرير: نسمة محمود
